Solution-focused toxicological risk assessment and medical device biocompatibility consulting
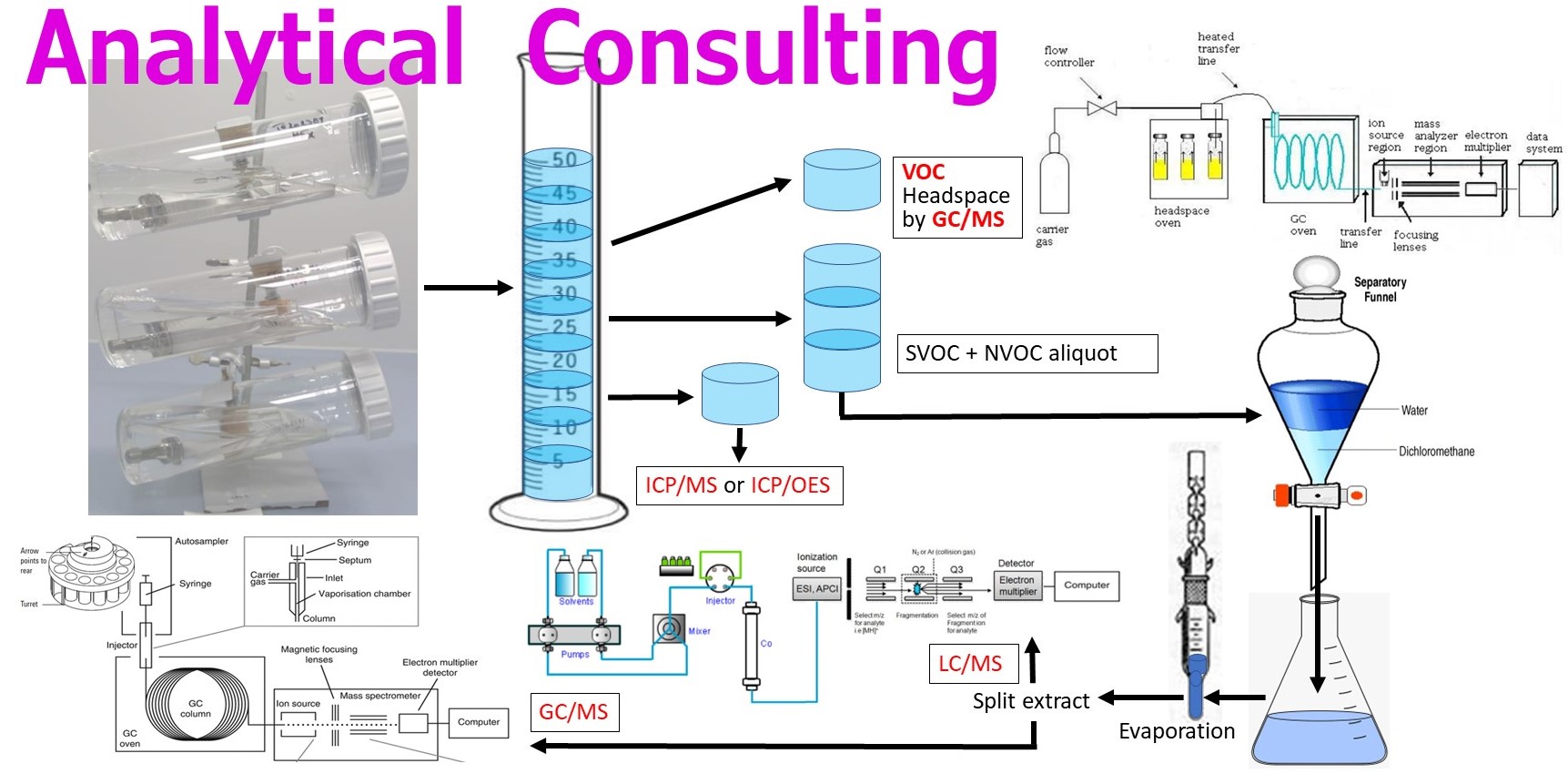
Analytical study design and protocol review
· Verify extraction solvent compatibility for polymers
· Exaggerated, exhaustive, or simulated use extractions
· Inhalation flow rate, sampling times, condensate test
· Analytical evaluation threshold: Verify LOQ and UF
· Optimize solvent reduction to minimize sample quantity
Review analysis report and mass spectral IDs
· Part 18 compliance: AET, exhaustiveness, & recoveries
· Omissions, artifacts, coelution bias, & misidentification
· Interpret spectra and conduct offline NIST library search
· Advanced instrumental analysis for unknowns >TTC
TDAC has 40 years of experience in planning, overseeing, and interpreting chemical characterization studies for extractables and has prepared over 300 proposals for extractable/leachable (E/L) testing of medical devices and drug product shelf life studies.
TDAC has experience with study design and oversight of E/L testing for a wide variety of medical products, including drug product container closure systems (CCS), prefilled syringes, drug coated balloon catheters, vascular, ventricular, and urinary catheters, cardiac valves, pumps, and stents, hemodialysis devices, in situ curing bone cements for hip and knee arthroplasty, in situ polymerizing cyanoacrylates for artificial embolic treatment of stroke, tissue adhesives, intravenous and subcutaneous drug delivery systems, transdermal patches, intraosseous needles, breast implants, wound dressings, tissue scaffolds, craniomaxillofacial reconstruction implants, orthopedic and dental implants, surgical instruments, bioresorbable sutures, 3-D printed bone scaffolds, humidified breathing circuits, MDI/DPI inhalers, nebulizers, oxygen and nitric oxide delivery systems, CPAP devices, and endotracheal and nasopharyngeal tubes.
A critical factor to gaining regulatory acceptance of a toxicological risk assessment is proper E/L study design, which uses extraction solvents compatible with the device raw materials and extraction methods that are consistent with recommendations in ISO 10993-18. For example, exhaustive extractions are recommended for initial chemical characterization studies for permanent implants, whereas release kinetics extractions can be used with follow-up studies involving targeted compounds that displayed high levels in the initial study, which may occur with soft polymers (e.g., silicones), resorbable materials, or where there are reactive leachables, such as in situ curing polymers. Fluid path extractions are appropriate for some externally communicating fluid delivery devices. Note that US FDA generally requires separate extractions of any device which directly contacts body tissues using polar, nonpolar, and mid-polar solvents, while regulators from other regions (such as Europe) may only require polar and nonpolar solvents.

For some polymeric devices, a preliminary solvent compatibility test may be recommended to ensure that significant degradation of the test article does not occur during contact with the extraction solvent at elevated temperatures. ISO 10993-18 warns that material degradation of a medical device may not be considered representative of clinical conditions, and in such cases the E/L study could generate an unrealistic extractables profile. In some cases, suitable solvents can be confirmed by examination of polymer solvent compatibility charts and solvent polarity tables; however, in other cases, particularly for soft polymers, a preliminary extraction or incubation of the test article in several solvents may be necessary. In addition, some medical devices may contain adhesives which might dissolve and cause disassembly of components during extraction.

For the analysis of drug product packaging/delivery components or prefilled packaging/delivery systems, ICH Q3E provides draft guidelines for conducting extractables testing under exaggerated conditions for the empty packaging components or delivery system. The results of the extractables study may be evaluated in a TRA to determine whether there is a need to develop a validated target compound method for testing for certain leachables that could be released in the presence of the drug product formulation. Either a method suitability study (MST) or a method validation study may be performed to develop a suitable and accurate analytical method which is able to detect trace leachables in the presence of the drug product solution. Following ICH Q3E, a quantitative leachables study may be performed to assess potential leaching for selected compounds of toxicological interest identified in the extractables study.
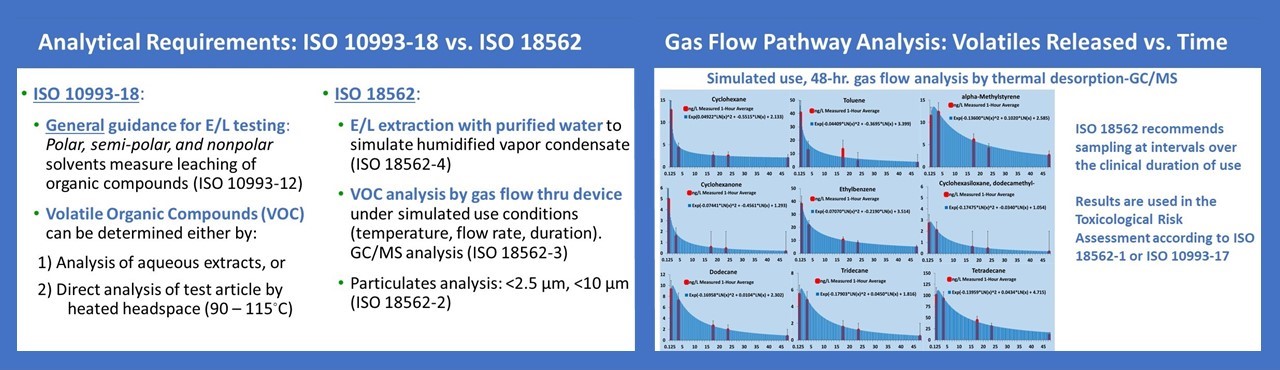
For the analysis of medical devices which are used for delivery of gas to the patient’s lungs, ISO 18562 calls for analysis of volatile organic compounds (VOCs) which are collected during a simulation experiment where gas is flowed through the device to measure the extent of off-gasing. During the simulation experiment, air samples are collected using either stainless steel canisters or solid sorbent collection tubes at several time points so as to bracket the VOC emission rates that occur over the timeframe of clinical use and allow an estimate of the maximum rate of release for each substance. ISO 18562-3 also notes that, if formaldehyde and other carbonyl compounds are identified during the VOC analysis they become target compounds and the analysis shall conform to ISO 16000-3 and ISO 16000-4. In addition, ISO 18562 requires an extractables study using purified water for any device that is indicated for use in a humidified gas pathway. Since the amount of water vapor condensate that could be inhaled by a patient varies with the specific conditions of use for a particular device, it is difficult to estimate what volume of solvent is representative of patient exposure. For this reason, an experimental simulation to estimate moisture condensate generation during clinical use is sometimes performed following ISO 9360-1. Lastly, under ISO 18562, inhalation pathway devices also require testing to measure particulates <2.5 µm and <10 µm.
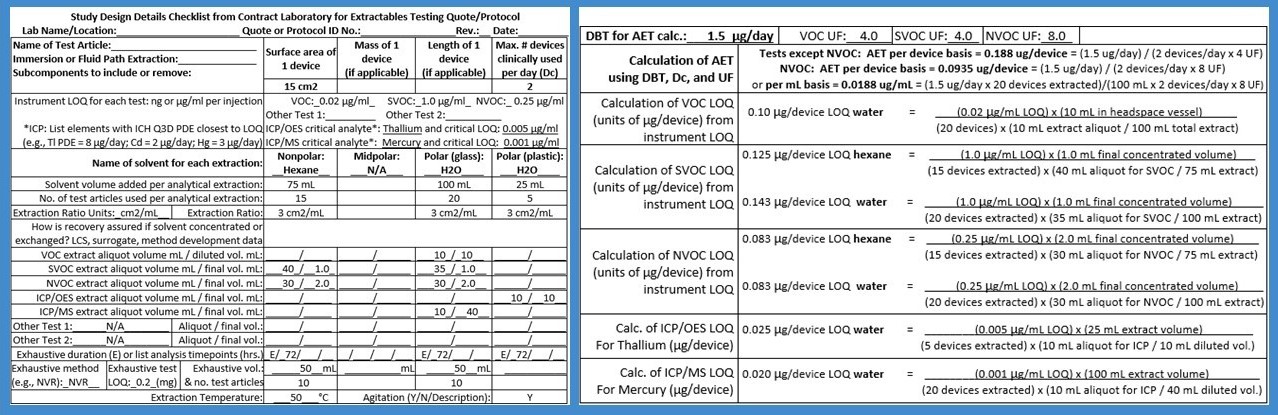
Verification of the Analytical Evaluation Threshold (AET) is advised prior to conducting E/L analysis for organic compounds, including VOCs, semivolatiles (SVOCs), and non-volatiles (NVOCs). The AET is defined as the minimum instrument detection sensitivity needed to ensure the ability to detect compounds of even the highest toxicity, while accounting for the possibility that some compounds may display a relatively low detector signal response. The AET is based on the Dose-Based Threshold (DBT), also known as the Threshold of Toxicological Concern (TTC), which represents an allowable daily exposure level that is sufficiently low to assume the absence of toxicological risk for nearly any chemical of a given class. The AET incorporates an analytical Uncertainty Factor (UF) to account for the range of compound-specific sensitivities that are associated with each type of instrument detector. Compared to GC/MS, LC/MS has a relatively wide range of analytical UFs.
All analytical methods used in E/L studies must be able to achieve a detection limit (i.e., Limit of Quantitation) which is less or equal to the AET, so that the reported data can be considered usable for the purposes of toxicological risk assessment. In other words, there should not be any risk of false negatives; i.e., failure to detect a compound above a TTC but below the detection limits, which would imply an uncertainty with unknown toxicological risks from clinical use of the medical device. The TTC is of key importance because when no published toxicological data exist for an identified compound or an unknown reported substance, the toxicological risk assessment may be based on a worst-case assumption of toxicity using a TTC. The use of a TTC applies only to organic compounds. The most stringent TTC for a device intended for long-term use (>10 years to lifetime) is 1.5 µg/day. Thus, the analysis of implantable devices must achieve a detection limit equivalent to (1.5 µg/device) / (UF). If detection is not feasible at this level, then potential options should be considered, such as utilizing a higher extract concentration factor, extracting a greater number of test articles, or using a different instrument with a more sensitive detector system. For devices intended for shorter periods of clinical use, the AET is based on a less stringent TTC, such as 10 µg/day for daily exposures from >1 to 10 years, 20 µg/day for >1 month to 1 year, and 120 µg/day for ≤30 days.
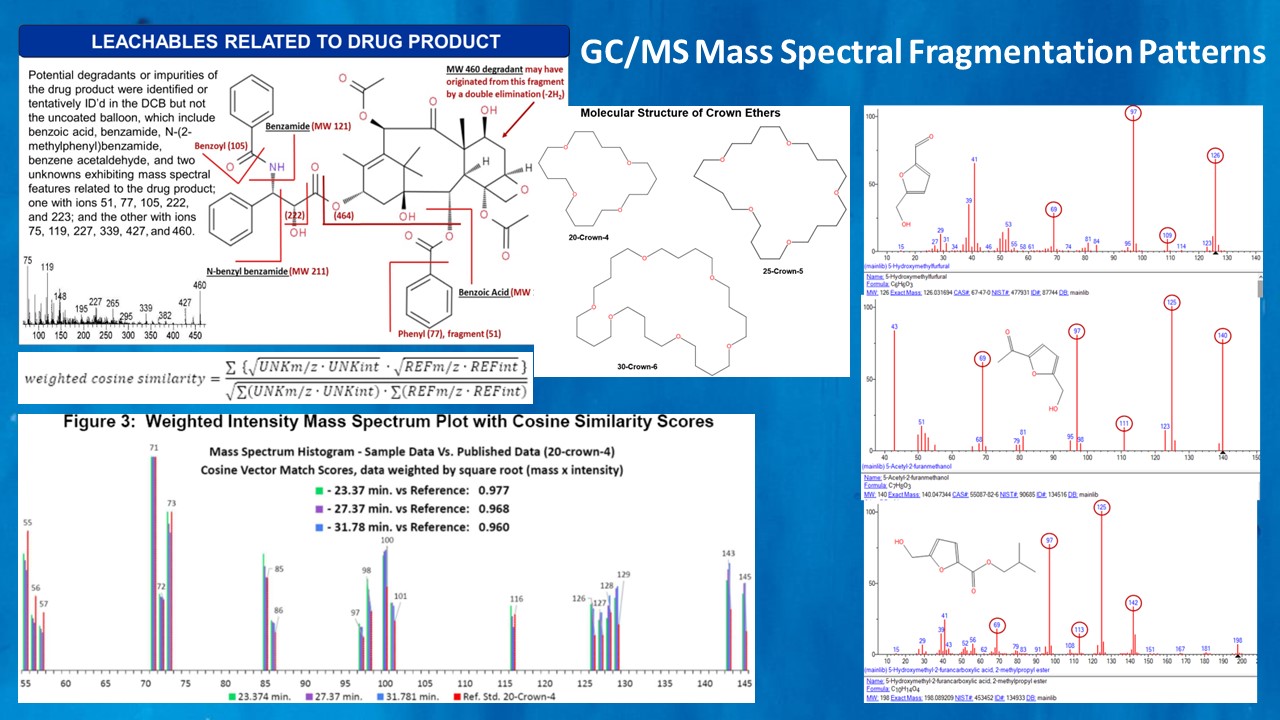
For compounds that cannot be identified (unknowns) or which were reported as very unusual or suspect compound identities, it may be necessary to request a re-evaluation of mass spectral data by a subject matter expert at the laboratory. TDAC has 40 years of experience in mass spectral data review and interpretation, and can frequently determine which compound identifications may be misreported, misidentified, or artifacts of laboratory contamination.
TDAC may request the laboratory provide a copy of suspect mass spectra in order to perform an independent library search of the NIST mass spectral database using a personal computer. In some cases, the sample mass spectrum may exhibit a non-unique match to several characteristic ions, and in such cases it can be worthwhile to conduct a reverse search of the candidate library match to see if another more common and more plausible substance exists which has a very similar mass spectrum.
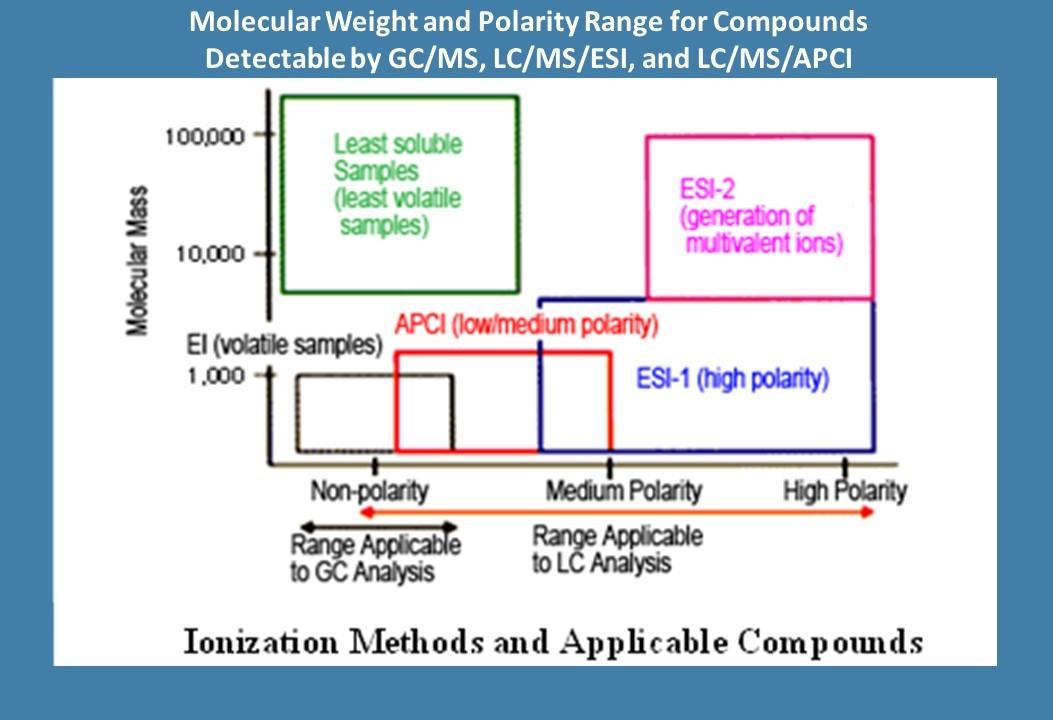
For E/L testing, LC/MS instruments should employ a High Resolution Accurate Mass Spectrometer (HRAMS) detector, which can include Electrospray Ionization (ESI) detection for polar extracts and Atmospheric Pressure Chemical Ionization (APCI) for non-polar extracts, and also univariate detectors such as UV or CAD, which may be used to measure other peaks which were not able to be detected and quantified by HRAMS due to low detection sensitivity. In addition, the LC/MS system should employ a two-stage ionization process with an initial soft ionization step used to generate the intact molecular ion or molecular ion adducts, followed by secondary ion fragmentation using Collision Induced Dissociation (CID), which generates more detailed mass spectra suitable for structural elucidation. At some laboratories, an automated instrument software algorithm is used to trigger the secondary ionization step, which is not foolproof and occasionally may cause peak detection criteria to fail so that secondary ion fragmentation data may be inadvertently omitted for some peaks. This situation requires careful data review and may require sample reanalysis if the data collection could have missed peaks greater than the AET.