Solution-focused toxicological risk assessment and medical device biocompatibility consulting
Biological Evaluation Plans (BEP)
· Gap analysis to review usability of legacy biocompatibility test data
· Study design per ISO 10993-1, optimize endpoint overlap with 10993-18
· Strategic portfolio test planning, group similar products/components
· Selection of appropriate GLP methods; review/approve CRO protocols
Biological Evaluation Reports (BER)
· Review passing/failing tests, assess deviations & unusual observations
· Test failure root cause analysis, formulate rationale or follow-up test plan
· Summarize biological testing and extractables TRA in BER
· Formulate conclusions on safety, uncertainties, and risk management
Strategy Optimization (biocompatible materials, predicates)
TDAC has experience designing biocompatibility studies and preparing biological evaluation plans (BEPs) and biological evaluation reports (BERs) for new product development projects, EU MDR remediation submissions, manufacturing process or material change evaluations, and CAPA remediation. TDAC has performed gap analysis of legacy biocompatibility reports dating from 10 to 25 years ago to assess whether such data would be usable for a new regulatory submission based on comparison to the requirements of current ISO standards.
TDAC has developed efficient product portfolio testing strategies which can limit the scope of testing to representative devices which are grouped by material composition, manufacturing process, and ISO 10993-1 tissue contact category. Careful consideration is given to selection of appropriate methods that will provide robust evidence to assess data usability; for example, the MTT test can provide numeric cytotoxicity scores for several extract dilutions, which can support a rationale for biological safety in cases where the undiluted extract is shown to be cytotoxic, but not the diluted extract.
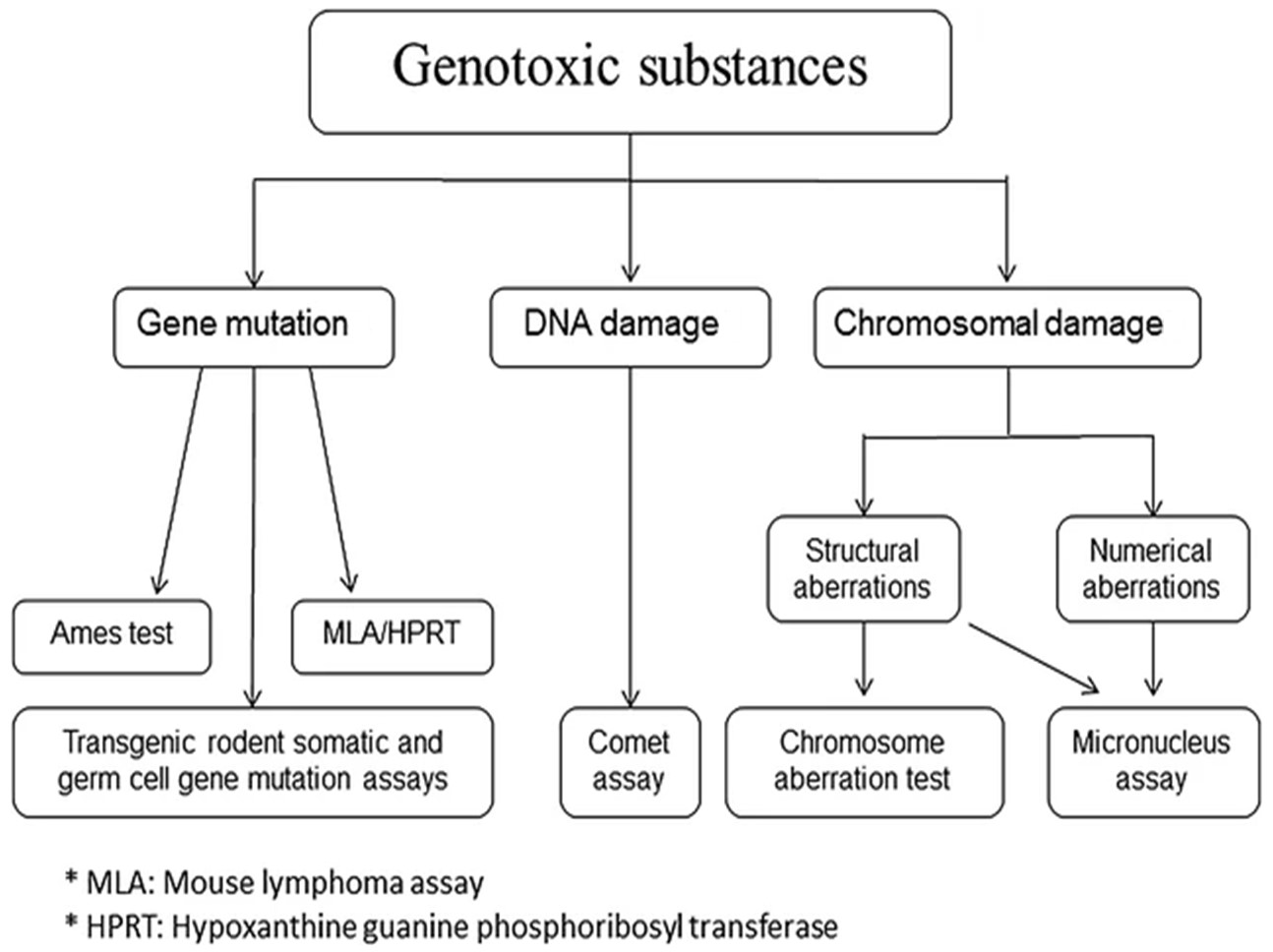
For genotoxicity testing of medical devices conducted in accordance with ISO 10993-3 and ISO 10993-33, a combination of the bacterial reverse mutation (Ames) assay and a mammalian test for genotoxicity may be recommended. In the event that one or both of these tests are positive, a follow-up in vivo genotoxicity test is recommended (such as the in vivo mouse micronucleus test), which may yield more relevant information as to potential genotoxicity under the conditions of clinical use. Genotoxicity testing is sometimes conducted only after E/L chemical characterization reports extractable compounds which are unable to be identified by GC/MS or LC/MS at levels above a stringent genotoxicity TTC.
TDAC gives careful attention to study design parameters associated with animal implantation testing. An appropriate implantation study protocol should justify the selection of a tissue implantation site, include monitoring of all required biological endpoints, and prescribe macroscopic and microscopic histopathological examination of tissues and organs to be evaluated at specific sacrifice intervals. Also, the test article implant size must account for a scaling factor to ensure the validity of extrapolating from animals to humans. The CRO selected to perform the study should have demonstrated experience in conducting similar implantation surgeries using the same animal models, and caution must be exercised because the surgical technique may profoundly influence the result of any implantation procedure.
To Test or Not to Test: Testing for some biological endpoints may be waived based on completion of chemical characterization studies along with an acceptable toxicological risk assessment. The characterization of extractables and leachables (ISO 10993-18:2020), together with a toxicological risk assessment (ISO 10993-17) may be utilized in lieu of conducting biological tests for subchronic and chronic systemic toxicity, genotoxicity, and carcinogenicity. To this effect, ISO 10993-1:2018, Section 4.3 states, “Chemical characterization with an appropriate toxicological threshold can be used to determine if further testing is needed (see Annex B, ISO 10993-17 and ISO 10993-18).” Section 6.3.1, Subpart b) also states, “The choice of test procedures shall take into account: … 4) that certain biological tests (i.e. those designed to assess systemic effects) are not justifiable where the presence of leachable chemicals has been excluded (in accordance with ISO 10993-18), or where chemicals have a known and acceptable toxicity profile, allowing the safe use by evaluation in accordance with ISO 10993-17 and risk assessment in accordance with ISO 14971.”
Selecting an appropriate biological testing battery requires professional judgement and is not a simple check-box exercise. Animal welfare guidelines should be considered to minimize animal testing. Some examples of typical strategies and rationale applied to justify waiving certain biological tests for tissue implants are given below; however, in some cases, novel materials may require more comprehensive testing.
- Section 4.4 of ISO 10993-2:2006 states, “For the purposes of the ISO 10993 series, animal tests shall only be deemed to be justified … when no suitable scientifically validated test method not involving the use of living animals is reasonably and practically available; and when relevant reduction and refinement strategies have been identified and implemented including, if appropriate, obtaining test data from manufacturers and suppliers, and literature searches for toxicity and biocompatibility data.” In addition, Section 4.4 states, “Testing is usually not necessary when sufficient information is already available to perform a risk assessment of the material and/or the medical device … biological testing is usually not necessary, if material characterization (e.g. physical and chemical) demonstrates equivalence to a previously assessed medical device or material with established safety.”
- Section 4.10 of ISO 10993-1:2018 states that, “The biological evaluation shall take into account preclinical tests, clinical investigations, post-market experience from similar medical devices or materials, and other relevant information.” This information can be leveraged as described in Section 4.11: “Where recommendations for endpoint assessment per Annex A are different from prior published versions of this document, a history of safe clinical use can be used to document why additional testing on a commercially-marketed medical device is not needed.”
- Regarding the relevance of testing for material mediated pyrogenicity (i.e., non-endotoxin related), Annex G of ISO 10993-11:2017 states, “It is not necessary to test all new medical devices. However, substances that have previously elicited a pyrogenic response, and/or new chemical entities where the pyrogenic potential is unknown should be evaluated for material-mediated pyrogenicity.” Annex G lists substances that are currently known to elicit a pyrogenic response, including endogenous pyrogens, prostaglandin, inducers, drug substances that disrupt thermoregulatory centers, organic chemicals that act as uncoupling agents of oxidative phosphorylation, naphthylamines, bacterial endotoxins, neurotransmitters, and certain nickel salts.









